|
|
| |
nmrshiftdb2 Help
|
|
|
| |
The main functions of nmrshiftdb2 (search, predict, submit, review) can be accessed via the tabs on top of the main page. Registration is necessary if you wish to contribute to the database; it is not necessary if you just want to use the Search and Predict functions. To register, simply click the "Create new account" button, fill in the form, click the "Do you want to become a contributor?" field as appropriate and submit the form. After successful registration you will receive an Email which provides a link which you must visit for confirmation. You can then officially log in to nmrshiftdb2. When logging-in, you can choose to save your user data in a cookie. This means you will be automatically logged-in when you visit this server in the future. The cookie will be deleted when you explicitly log-out; just closing the browser will keep the cookie. Note that this is inherently insecure when other people use your computer with your identity. Only use this when you are sure that you have a secure login on your computer. nmrshiftdb2 will keep your personal data secret. There are the following exceptions: Your user name will be visible with spectra you entered. nmrshiftdb2 reviewers can read your real name and email adress, if you did submit anything to nmrshiftdb2. The rest of your personal data can only by read by the Administrator and should not be used except for verifying that you are a real person. The bunch data available on Project Homepage at SourceForge do not contain personal data.
nmrshiftdb2 servers log the queries entered by users. These queries are not published and will not be available to anybody except the Administrator. Statistics showing the distribution of queries to query types are compiled and may be published, but they do not contain any individual queries. Logging data may also be used for research purposes, but only results of this research will be published. Note that the privacy policy given here only applies to the server(s) reached via the www.nmrshiftdb.org URL (the nmrshiftdb2 data project). The software developers can not give any guarantee that other sites running the nmrshiftdb2 software (which is open source) comply with this policy. If you are interested in becoming a reviewer for new submissions, please send an Email to the administrator. For registered and logged-in users the individual Search History and the Structures History will be saved after each session and reloaded automatically at the next login. Registered users also see a link "Show all my contributions" on the search page. This displays all contributions plus their status and the assigned reviewers. We also offer the possibility to registered users to download a list of their latest contributions for inclusion in their homepage. A link like http://www.nmrshiftdb.org/NmrshiftdbServlet?username=name&nmrshiftdbaction=exportlastinputs&numbertoexport=5 will give an html fragment (not complete page). Here username is your username and numbertoexport determines the number of structures (optional, default is 4). Connections should be made by secure https protocol automatically. Trusted certificates are provided. If you get warnings relating to security, check them carefully to make sure there is no problem with your connection. On standard systems, the web page should display with the appropriate security symbols of your browser being visible. The Search user interface is started by clicking the "Search" button near the top of the nmrshiftdb2 window. This action opens the Search pane. The default pane only contains the most important search options; if you want more and more complicated options, choose one the "Switch to expert search mode" links. There are a variety of search algorithms described below that allow the user to search for database records on the basis of a list of chemical shifts, chemical name, molecular weight, molecular structures, or other text descriptors (properties) and keywords. Since nmrshiftdb2 contains both measured and theoretically calculated spectra, check box options are provided to specify whether subsequent searches should consider only measured, only calculated, or both types of NMR data. A Spectrum Search, i.e., the search for a given set of chemical shifts defined by an input list, can be done in two different ways. The shifts must be entered in "Input Field" (one resonance per line). The format is like this:
shift (ppm)
shift (ppm); intensity (a.u.)
(intensity is optional)
Example:
56.2;.42
64.1;.6
125.6;.3
The search types are: Subspectrum Search (match all Input as subset of Record): a similarity search which looks for spectra in the database for which a subset of the total NS resonances exhibit chemical shifts equal to or close to the NI values defined in your input list. Matched spectra must contain the same or a larger number of resonances compared to the input list, i.e., the input spectrum can be a subspectrum (NI<=NS). For each spectrum found a similarity factor is calculated, which is a measure of how closely your input data are matched by the relevant chemical shifts in the database spectrum. A Hitlist of spectra is returned, in the order of decreasing similarity. The similarity measure is 100% if all NI peaks in the input list can be exactly matched by a subset of peaks in the database spectrum, regardless of NS. Complete Spectrum Search (match complete Record to Input List): a total similarity measure is calculated by comparing all NS resonances of the database spectrum with the NI chemical shifts in the input list. A max. similarity of 100% is possible only when NS = NI (identical spectra); otherwise, when NS is not equal to NI, the max. similarity measure is reduced to the factor 100*(no. of matched peaks)/(no. of matched + unmatched peaks). Thus, the similarity measure reflects how well each peak in the complete database spectrum can be paired with one value in the input list on a one-to-one basis. The Hitlist can contain not only matched spectra with NS = NI but also spectra which contain the input spectrum as a well-matched subspectrum (NINI). This procedure is slower than the simple similarity search.
Example: the Input Spectrum is [24, 45, 66, 88] (four chem. shifts in ppm). Subspectrum Search: Only matching spectra will be found which contain at least 4 resonances. For example, the spectrum [24, 46, 65, 89, 95] contains the input spectrum as a subspectrum with a similarity measure of ca. 97%. Complete Spectrum Search: The following matching database spectra would be found: spectrum [23, 47, 64, 87]; similarity ca. 96% (4 of 4 resonances matched) spectrum [26, 45, 65, 90, 110]; similarity ca. 75% (4 of 5 resonances matched) spectrum [25, 45, 66]; similarity ca. 72% (3 of 4 resonances). A spectrum search requires an input spectrum as a list of chemical shifts and (optional) relative intensities (they will be scaled to the 0-1 range automatically). One resonance per line must be entered in the Text Field provided, either manually or by Copy/Paste from another document. Both decimal point and decimal comma are allowed; shifts and optional intensities must be separated by semicolon (;) or colon (:). You can also define a spectrum type to search with the "Spectrum type to search for" drop-down box - this will limit searches to spectra of this type. Example of an input spectrum with intensities: You can also add a multiplicity identifier (S, D, T, Q) at the end of a line. This means that only signals will be considered as matching the multiplicity of which are the same as the given multiplicity. Notice that other multiplicities than capital SDTQ are removed, since this function is only useful with carbon spectra, where multiplicities are reliably available. Other spectra, like 1H spectra, do not have reliable multiplicities given. After the input spectrum list has been entered and the search algorithm has been chosen, click on the "Search by spectrum" button to execute the search. Instead of typing the spectrum you can also upload a jcamp file. Choose it via "Browse...", check the search type and hit "Search this spectrum" to execute the search. If the file contains a peak table, this will be used, if not, a peak picking is performed. The dropdown box "Spectrum type to search" determines the nucleus of the spectra searched. 13C is default. Note that there is also a general type selection for more advanced users. If you use this, it must at least contain the type you choose for the spectrum search, else you will get empty results. You can also do a 13C/1H correlation search by chosing the option "13C/1H correlation" and entering a shift table like this: This searches for molecules where 56.2 and 2.2 are on neighbouring carbon/hydrogen atoms, as well as 64.1 and 2.6. The error message "Your input included unallowed characters" means that your input was not correctly formatted (non-numerical characters, improper separators, empty lines). Correct your input and submit it again. The results are displayed in the "Results" table. The molecular structures associated with the matched spectra are displayed together with the computed similarity measures. The matching performed for the similarity calculation is also displayed. If a particular molecule appears more than once, then more than one spectrum is available in the database (e.g. different solvents). For more information about browsing the results see Browsing the Results. In case of a spectrum search you have two additional informations under teh spectrum: Firstly the threshold, which means how much percent are contributed maximally by one shift and the unused signals. Unused means they did not match anything. By using the "Browse all spectra" link you can simply browse through all structures and their spectra. Also here you can use the spectrum type selection. In order to search the database for molecular structures, it is first necessary to create or input a structure in an appropriate encoding format which nmrshiftdb2 understands. You can either upload a file in common formats (valid formats) via the "Browse" button or activate the Java based version via the  symbol. You can then use the JChemPaint applet to interactivly paint structues. You can also import a file by Copy & Paste via the "Import text by copy &paste button". symbol. You can then use the JChemPaint applet to interactivly paint structues. You can also import a file by Copy & Paste via the "Import text by copy &paste button". You can enter stereochemical configurations by using solid (up) and dashed (down) wedged bonds. These will be used for total structure search, but currentliy not for the substructure search. If the total structure search finds nothing with respecting stereochemistry, it "falls back" to not using stereochemistry. Once the desired structure has been drawn or loaded into the Marvin Sketch window, the type of search to be performed can be selected by clicking the check box next to "substructure search" (look for structures containing the input structure), "similarity search" (look for structures with a similar substructure) or "identity search" (look for structures identical to the input structure). Note that the similarity search will always be a superset of the substructure search. Click on "Search by structure" to start the search. The results are displayed on the "Results" tab. For more information about viewing results see Browsing the Results. The drawn structures are collected in the Structures History. You can return to Marvin to make changes in the input structure or clear the sketcher window with the "Clear" command to prepare for new input. In addition to the molecular structure and numerical spectroscopic data, each database record also contains various text items called Properties (e.g., chemical name, molecular formula, spectrometer frequency, solvent, etc.). With the dropdown list box you can choose the text field to be searched (some are available in in expert mode only). A particular search is "Chemical Name (with Pubchem name resolution)", here the name entered is looked up in Pubchem and the actual search done using the InChI found. Since Pubchem is very comprehensive, this should find structures by any reasonable name. After selecting the appropriate search algorithm (see below) and entering the desired text search string, click "Search" and the results will be displayed on the "Results" tab. For more information about viewing the results see Browsing the Results. There are three possible algorithms for the Properties Search. Exact Search: All records will be found where the contents of the selected field exactly match your search string. Fragment or Regular Expression Search: You may enter a specific text substring (fragment) and all records will be found which contain this substring anywhere within the selected text field. Alternatively, you may perform a more sophisticated search using Regular Expressions. For details about regular expressions, there are many good explanation in the internet, e. g. here. Fuzzy Search: This is a clever combination of the Soundex algorithm, the Levenstein Distance Algorithm (LDA), and a substring search. Records will be retrieved for which the selected Text Field contains the search substring, not only on the basis of alphanumeric text but also on the basis of similarity when spoken as American english (useful when spelling is uncertain). The results are presented in the order or decreasing similarity. Due to the fuzzy nature of the algorithm, a large number of records may be found with small search strings. The Fragment Search is more efficient if the correct spelling of the search text is known.
Please note that a Chemical Formula search is a special case of the Properties search. The Regular Expression/fragment search is not allowed. The Exact Search will retrieve all molecules whose formulas contain the same elements and numbers given in your search string, regardless of the ordering of the elements in the formula. If no number is given, one is assumed as default, so CH4 is equal to C1H4. Correct capitalization is necessary since e. g. Si is different from SI. The Fuzzy Search requires an input string with the correct chemical elements but the associated number for each element can be given either as an exact number n, as a range of values n-p, or as a wildcard *. For example, the search string C5-7H*Br2 will find all molecules containing 5 to 7 carbons, 2 bromines and any number of hydrogens. With "Chemical formula" respectivly "Chemical formula (with other elements allowed")" you can spedify if other elements apart from the elements given are allowed or not. With additional elements, the mentioned query will find all molecules containing 5 to 7 carbons, 2 bromines, any number of hydrogens and any other elements. In contrast, only Chemical formula will find all molecules containing 5 to 7 carbons, 2 bromines and any number of hydrogens and no other element. The search by multiplicities works similar to the chemical formula search, but instead of element symbols you need to use symbols for multiplicities. A regular expression search is not possible. Example: S*D2-3T4 will find all molecules containing any number of singlets, two to three doublets, four triplets and no quadruplets. Note there are two multiplicity searches: The real multiplicity search, which looks for multiplicity identifiers as entered by the contributor and the potential C13-multiplicity search. This looks for multiplicities as derived from the H count, no matter what the actual data are. Possible symbols are S, D, T or Q for singlets (no H), doublets (one H), triplets (two H) or quadruplets (three H) respectivly. You can search for molecules whose molecular weight falls within a range by giving the range like "100-200". Click "Search" and the results will be displayed on the "Results" tab. The search by keyword/category (expert mode only) is similar to the text string search for molecule/spectrum properties described above. However, in this case you can choose from a list of defined keywords/categories, and you can select multiple keywords/categories by holding down the "Ctrl" key. With the "Total keyword search" you search for the complete keywords, with "Keyword fragment search" all keywords containing fragments of the choosen keywords, split by blanks, will be found. Example: Keyword fragment search for "abietic acid" gives all molecules with keywords containing "abietic" or "acid". Here you can search for spectra with a certain measurement or calculation condition (expert mode only). All search procedures can be restricted to database records containing only measured or only calculated spectra, as desired, by clicking the appropriate check boxes (expert mode only). A selection will be valid till you change the settings, not just for one query. All search procedures can be restricted to certain spectrum types (expert mode only). Check the types you want to search for by clicking the appropriate check boxes. A selection will be valid till you change the settings, not just for one query. Here you can also determine if unreviewed spectra should be used for predictions. By default, only reviewed spetra are used. If you uncheck this box, also unreviewed get used. This can be used if you entered several spectra, which are not yet reviewed, but already want to try how this influences predictions. A selection will be valid till you change the settings and will influence the predict function as well as prediction during submit. If the server is a Lab server (not the public servers) all searches can be restricted to submissions from the lab only. A selection will be valid till you change the settings, not just for one query. Futhermore, there is a setting "Use .SHIFT REFERENCE in jcamp files". If this is set, SHIFT REFERENCE in JCAMP files will be used for calculating ppm. By default, it will not. We found many files contain this, but have actually already done the shifting. The results of each search, i.e., the matching records retrieved from the database, are displayed in the "Results" pane in the form of molecular structures. Up to ten records can be shown per page and up to ten pages are accessible via the numerals at the top of the pane. If necessary, further blocks of ten pages can be accessed by clicking on the appropriate Arrow symbol next to the page numbers. The details of first structure on a page are shown directly. To view the details of a particular record, click on the structure of interest in the Results pane. Instead of choosing a structure you can also browse in a page with the "Browse forward"/"Browse backward" buttons. In the right half of the Results pane the spectra for this structure are shown. If there was a set of spectra (a "dataset") measured with the same sample submitted for the structure there will be several tabs with the name of the dataset(s) visible at the top, with the spectra not in a dataset being in the "General" tab, otherwise there will be no such tabs. For each spectrum three tabs are shown: - Spectral Data (chosen by default): The structure is shown, together with either an image of the spectrum, if any exists (this can be enlarged via the "Show spectrum in full size" link), or a schematic stick spectrum otherwise, and a table of atom numbers and assigned chemical shifts. By clicking on the header of the atom number or shift column, you can sort the table by any column in ascending or descending order (indicated by ⇑/⇓). In a lighter color other spectra of the same type are given for compararision. On top of a structure display, there is a symobl () for switching to Java-based display. In Java mode, the correlation between atom position in the structure and chemical shift value can be viewed by hovering with the mouse over a shift value in the data table. The corresponding atom in the structure and the peak in the stick spectrum will be highlighted. Pointing with the mouse to a peak in the spectrum causes the shift value to be displayed. You can zoom or expand a desired spectrum region using the mouse to position two lines marking the left and right limits of the expansion. Click "Zoom to original size" to return to the full display. On top of a structure display, there might also be a symobl (
 ), whch shows that there are 3D-coordinates available for this structure. They can be viewed in Java-mode. The structure shown in the Details window is shown as a graphic or, if you are in Java mode, via the JChemPaint or JMol applet, depending on if you have got 2d- or 3d-coordinates. The structure in JMol can be manipulated (scaling, translation, rotation) via a menu which can be accessed by pressing the right mouse button while in the structure window. ), whch shows that there are 3D-coordinates available for this structure. They can be viewed in Java-mode. The structure shown in the Details window is shown as a graphic or, if you are in Java mode, via the JChemPaint or JMol applet, depending on if you have got 2d- or 3d-coordinates. The structure in JMol can be manipulated (scaling, translation, rotation) via a menu which can be accessed by pressing the right mouse button while in the structure window. - Additional Data: This gives text information about Structures (names, weight etc.) and spectrum (measurement conditions etc.).
- Download: The download tab offers the viewing and download of structure and/or spectrum in various formats. Choose a format and click the "Request" button.
If there are several spectra of one type, they can be viewed by vertical scrolling. Order is by spectrum type. Calculated or predicted spectra come after measured spectra. Note: the atom numbers (labels) may appear to be rather arbitrary; they are defined by the numbering scheme generated by the structure editor (depends on the order of drawing operations) or present in the mol file used as input. The numbering will most likely not correspond to IUPAC or any other systematic scheme. This is a major inconvenience when comparing data for a variety of related structures. If you are a registered contributor, you can also add a new spectrum for an existing molecule in the database by clicking the "Add a spectrum" button. A database record of particular interest can be added to a "favorites" list by clicking the "Bookmark" button. The name of the structure will then be displayed in the bookmarks section at the top of the page and you can recall the structure by clicking it's bookmard name. The link described as "Copy molecule link" on the bottom of the page can be used as an external link to exactly this dataset without searching for it. For copying it, use the context menu (right click with the mouse) of your browser. The search history window in the right half of the "Search" pane will keep a history of all searches performed in the current session (log-in) or, for registered and logged in users, for up to 50 searches over the current and past sessions. Single or multiple searches shown in the history window can be repeated by marking one or more check boxes and clicking the "Repeat Search" button. If multiple searches are selected, you can choose if they will be "and" or "or" combined. The "and" option menas you get all datasets fullfilling all conditions at the same time, "or" means they fullfill at least one condition. "And" should be used if you want to combine different categories, e. g. a solvent and a chemical formula. "Or" will typically be used to combine several possibilities of the same category, e. g. several solvents. You can also choose the "not" option. This will put a not in front of all conditions and therefore select everything not fullfilling the conditions. This means it will be the "rest" of the database. In order to submit data for nmrshiftdb2, you need to be a registered user. Please do not use the "back" and "forward" button of your browser while doing a submit. To do a submit, click on the "Submit molecule" tab. You will see a timeline with various stations where you can enter data. We recommend to start from left to right, but you can always go to any station by clicking on it. If you finish one station by clicking the prominent blue button you get to the next station. Green means the data for this station have been correctly filled in (if a station is green from start, it is optional). What you can and have to enter depends on the type of spectrum you want to submit. There are the following possibilities: - 1D spectra with peak list: These are the standard one dimensional spectra like 13C or 1H. Here you have to submit a peak list and ideally assign the peaks. A spectrum image will be created from the peaks and displayed, if no other image is available.
- Other 1D spectra: For these, e. g. NOE specta, no peak list is needed. Instead a spectrum image must be uploaded, which will be displayed. An unassigned peak list can still be entered, if wished.
- 2D spectra: For these no peak list is needed. Instead a spectrum image must be uploaded, which will be displayed. An unassigned peak list can still be entered, if wished.
The stations on the timeline are the following: Choose experiment (ony on lab servers, not relevant on the public instance): Here you can choose one of your orders. If you do so, data from the order (structure, conditions) will be used to populate the submit. If you skip this, you do a normal submit. Enter molecule: Here you enter the structure (if you add a spectrum to an existing structure, this is already filled in). You can either upload a file in common formats (valid formats) via the "Choose file..." button or activate the Java based version via the symbol. You can then use the JChemPaint applet to interactivly paint structues. You can also import a file by Copy & Paste via the "Import text by copy &paste button". You do not need to (but you may) draw implicit hydrogens. After you drew or imported your structure, submit it by clicking the blue "Submit molecule" button. Your structures are collected in the structures history. If you want to change the structure later, just hit "Enter molecule" again. You can upload mol and sd files from MNova (sd files only work in MNova 11 or higher) or mol files from ChemDraw with labels via the "Upload" facility below the structure editor. The structure and custom numbering, if in the file, will be read. From the sd file the shift lists will also be read. If there is more than one spectrum in the files you can choose which one you want to submit in the next step ("Set spectrum type"). If you want to submit more than one spectrum you can repeat the upload after submitting a spectrum. Set spectrum type: Here you enter the type of the spectrum (13C, 1H etc.) you want to submit. Add shifts: Here you enter the signals of your spectrum. For 1D spectra you need to give one signal per line, shift and intensity separated by ;. Example: The intensities are optional. They will be scaled to be in a range from 0 to 1, if they are not. After you input the spectrum, click the "Submit signals". You can add additional signals later by the same steps. You can also upload a jcamp file by hitting "Browse..." and choosing the file. If the file contains a peak table, this will be used, if not, a peak picking is performed. If the spectrum type (nucleus) is in the file, it will be used, else you also need to choose the spectrum type. If the spectrum type is 1H, you need to perform a partly manual peak picking. You will be presented with an applet containing all individual peaks. Most likely, these will contain multiplets, noise etc. You need to left click on the field and go over some peaks and release the mouse. Then the average of all peaks in this range will be calculated and added to the peak list on the right. If you submit, this peak list will replace the automatic peak list.For 2D spectra the format is slight different: The first two figures are the values on the two axis. The third gives the coupling constant and is optional. These peaks will not be processed in any way, but saved as given.Do assignment (for standard 1D spectra only): Here you have got one line per atom, showing a drop-down box for the shifts and the expected range for the shift. These predictions are calculated based on HOSE codes, as in the prediction function. Additionally there is a range for the signal given. In some rare cases the predicted value will be identical with one of the limits of the range. Blue figures indicate that your value is inside this range, red means outside. Choose a shift for every atom. In the "Atom identifier" column you can enter identifiers for the atoms (any character allowed) which will be used instead of the standard numbers. You should make sure identifiers are unique. If there is no identifier for an atom the number from the "Atom number" column will be used. You can also use this to renumber the atoms by using numbers as identifiers. Again the numbers should be unique. If you enter a spectrum for an existing structure you can chose an existing set of identifiers below the structure diagram. "Standard" means the numbers as they appear in the structure drawing. You can also edit an existing set by changing some identifiers. Shifts not assigned can be deleted in a later step or kept as unasssigned - this is also a way to get rid of mistyped shifts. After you assigned all signals, press the "Submit assignments" button. If you forgot to submit a signal, finish this step and go to "Add shifts" and then "Do assignments" again. If you have entered only one shift, it gets assigned to all atoms automatically and you may skip this step. You can also enter multiplicities for atoms (except for proton spectra, the default is calculated by the proton count of an atom) and coupling constants for every atom-atom combination. If you have not entered the shifts before this step, you can them enter here directly. Add miscallenous data: Please choose first if the spectrum is measured or calculated. Then enter the chemical names, any links to web-literature about the molecule and the CASNumber and autonom name of the molecule, if known (you do not need to do that if the molecule is already in the database). From time to time IUPAC and index names for molecules in nmrshiftdb2 will be auto-created, so you do not need to enter them. You can also enter literature and web pages about the spectrum. Further more, you can give keywords for the molecule and categories for the spectrum - either existing ones or new ones. Molecule keywords should be compund-specific, e. g. alcaloids. The spectrum categories, on the other hand, should refer to the spectrum, e. g. a certain institution they have been measured at. You also need to enter the conditions for the spectrum (required). Which conditions are needed depends on if the spectrum is experimental or calculated, which can bothe be stored in nmrshiftdb2. The experimental spectra are acquired by dissolving a small amount of a real world compound in a solvent. The solution is inserted into a strong magnet of a given field strength at a given temperature (typically around room temperature) and irradiated with strong electromagnetic pulses in the radio band. The electromagnetic response given by the molecule yields the NMR spectrum, which can - for the 1-dimensional case - be characterized as a collection of signals at a particular radio frequency, one for each atom of the chemical element type for which the experiment has been performed. While physicists use the SI unit "Tesla" as the correct unit for magnet strength, NMR spectroscopists traditionally use the resonance frequency of protons, which depends on the magnet strength, to denominate the strength of an NMR magnet. In magnets with a magnetic field strength of 11.75 Tesla, for example, protons resonate at 500 MHz. The parameters used by nmrshiftdb2 to characterize the experimental conditions under which a spectrum has been recorded are: Temperature [K] (example value: 298) Spectrometer Frequency for 1H [MHz] (example value: 500). Note that the frequency of the spectrometer is required, irrespective of the nucleus. This will be tranformed automatically. E. g. if you measure on a 500 MHz machine and the spectrum type is 13C, you enter 500 and 125 MHz will be displayed as "Field Strength [MHz]" in the database. Solvent (example value: CDCl3) Assignment method (example value: longrange Hetcor). If you are a labgroup user (not relevant for public servers), you will be presented with a list of possible experiments in your lab. The experiments which you click will be concatenated to form the assignment method. Alternatively, you can enter a text.
The calculation conditions focus on describing the quantum chemical computer program, which has been used to perform the calculation as well as the theoretical methods involved. There are a number of different ways to calculate an optimal molecular geometry and its associated NMR spectrum. It is important to list the terms commonly used for these methods together with the calculated spectrum in order to give users the opportunity to judge the quality of the calculated data. Since the qualitiy of a calculated nmr spectrum depends on the quality of the underlying molecular geometry, both the calculation method for the geometry as well as the method for calculating the chemical shifts have to be listed. At the time of this writing we restrict the type of calculated spectra which are allowed in nmrshiftdb2 to those calculated by so called ab initio methods, as opposed to semi-empirical and other methods, like HOSE code based or neural network based methods. The parameters used by nmrshiftdb2 to characterize the calculation parameters by which spectrum and molecular geometry have been calculated are: Program (example value: Gaussian98) Means "Quantum Chemical Program". The computer program by which the calculation has been performed (e. g. Gaussian 98) NMRMethod (example value: GIAO)
A methods described in the literature by which the Magnetic shielding tensors are calculated (e.g. GIAO for Gauge Independent Atomic Orbitals or CSGT for Continuous Set of Gauge Transformations) GeomModel (example value: B3LYP)
The model chemistry used for the geometry optimization, like RHF for Restricted Hartree Fock or B3LYP for Becke's frequentyl used DFT parameter functional. GeomBasisSet (example value: 6-31G(d))
The basis set used for the geometry optimization, like 6-31G(d) NMRModel (example value: B3LYP)
The model chemistry used for the shielding tensor calculation (possible values like in GeomModel) NMRBasisSet (example value: 6-31G(d))
The basis set used for the shielding tensor calculation (possible values like in GeomBasisSet) NMRStandard (example value: TMS)
Ab Initio calculations of magnetic properties yield shielding tensors, which are absolute physical quantities, whereas experimental spectra yield so called NMR chemical shifts which are given relative to the resonance frequencies of a given standard compound (usually Tetramethylsilane, TMS). Isotropic shielding values obtained by an ab initio calculation thus need to be subtracted from the isotropic shielding values obtained for a reference compound (usually also TMS). The calculation for the reference compound needs to be performed under exactly the same calculation conditions that have been used for the actualy compound in question.
Add literature items (optional): Here you may enter literature references for your data. Press "Submit literatures" once you have entered the literature items. Attach files: Here you can attach a JCAMP-DX, PDF, raw data or image file to your data. The file will be saved and displayed with the data and be available for download. Note that this file is saved independently from the peak list. If you used a JCAMP-DX file for creating the peak list, the file uploaded for the peak list will also be saved. You can remove the file using this function if you do not like your file to be included. The image file is mandatory for 2D and 1D spectra other then the standard ones. Final submit: Once you have entered all the data (the timeline is completely green) you finally submit your data here. You can choose to submit the data as private data (see Private Submissions ) and you can decide to submit a further spectrum for this molecule. If you choose this option, you will be taken to the start of the timeline with molecule-related data taken from your last submit. Click the "Write to database" button to finish your submit.
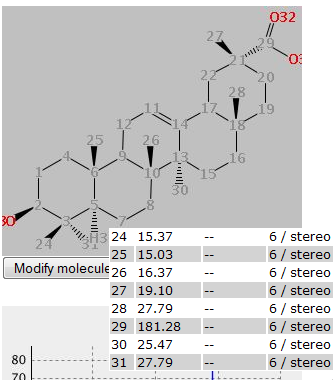
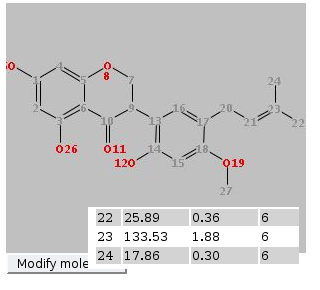
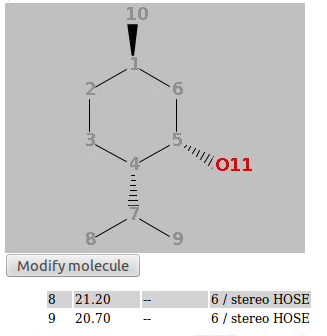
Your data will not be available immediately, but after they have been confirmed by a reviewer (you will get an email telling you who is your reviewer). To check the status of your contributions, use the "Show all my contributions" link on the Search pane. If for some reasons your submit is interrupted, you can continue that when you log in next time. You will see a blue box on top of the submit page where you can choose to continue or to discard the broken submit. If you are logged in, you can edit molecules you have entered. In the details portlet, you will see a button "Edit this spectrum". If you click this, you can follow the submit procedure and change anything you like. The changes need to be approved by the reviewer of the original spectrum. You can keep data you submit private and publish them later. This can be done by checking the checkbox "I want to keep this submission private" on the final submit page. This means that the data will not be visible to anybody except you. For you, they will be found by the normal searches and can be viewed via the link "Show all my contributions" on the search page or via your personal page (tab "My private submissions"). If you view the data, you will find the export facilities in the normal place and a button "Submit for review" under your spectrum. If you press this button, the dataset will be submitted for review and will be treated like any other submit. In the details view, you can also download your data in the same formats as other data. You can also edit your data and keep them in private status. In this way, you can enter your data while working on them and only publish them once you have the final data. When you view your private data there is a "Copy link for reviewer" option, where you can copy a link which enables access to this spectrum for a third party. The link contains a secret key and only works for this spectrum. Everybody who gets the link can view the specrum. Only you have access to the link and can decide to give it to others. A word on data protection here: Whilst we take any reasonable effort to secure our servers agains intruders, we cannot guarantee that there is no succesfull hacker attack. So we cannot take responsibility for loss or damage incured by your private data becoming known to third parties in any way. Reviews can only be done by registered reviewers. If you are one, you get notification by email about reviews assigned to you. In this email you find a direct link to the review page. You may also enter the id given in the email in the text field on the review page. In every case you get a graphical representation of the molecule and the spectrum. You will also find the signals assigned to an atom and the predicted signals and range, as during a submit. Decide if the data seems correct and click one of buttons at the bottom of the page. "Accept" means that data will be available for searches from now, "Reject" means spectra will be deleted from the databse, "Edit and accept" gives you the chance to change things before data becomes available, with "Abort" nothing happens and with "Send" you can send an email to the reviewer. With the "Ask the contributor to edit the contribution ?"-checkbox you can decide if the user should be asked to change something in the data. Nothing happens to the data, so you will need to review them later (you will be notified if the submitter has edited the data). If you see a "Return to editor" button, it means all reviews are assigend manually by an editor and you can decline to review this entry. After a review it may take up to an hour till data actually become available for spectrum and structure searches (other searches work immediatelly). Via the "Personal Page" link on top of the page, you can go to a list of all reviews assigned to you and not yet done. Here you can choose an unreviewed spectrum and go directly to the review page. As a logged-in reviewer, you can edit all data by clicking the "Edit this spectrum" button in the details portlet. You can follow the input procedure and change anything. A review is not necessary. nmrshiftdb2 can do an automatic assignment if you have a spectrum and a structure. This can help you in preparing data e. g. for a publication. To do so, you need to enter your molecule (about entering molecules see Submitting Spectra) and your spectrum as a list of shifts (ppm values) and to submit both. Your structures are collected in the structures history. The assignment is done by doing a prediction and assigning your shifts so that the similarity between the prediction and your spectrum is maximized. Therefore, the quality of the assignment depends on the quality of the prediction and the same limitations apply (see Prediction). You are then presented with the assignment in a table and a picture. If you hover over the table, an atom and its shift are marked. The table also gives the prediction for each atom and the difference of the prediction and your shift. Once the assignment is done, you actually have an almost complete nmrshiftdb2 dataset. You are therefore encouraged to submit your data by hitting the "submit your data" link above the table. During the submit, you can also correct assignment manually and add a literature reference. Submitted data can also be exported for publication. The Quick Check option offers a possibility to enter a 13C and a 1H spectrum as easily as possible and produce a quality measure for the spectra at a mouseclick. If satisfied the spectra can be submitted. A tutorial (en)/tutorial (de) on the Quick Check is available. You can upload mol and sd files from MNova directly (sd files only work in MNova 11 or higher). The structure and custom numbering, if in the file, will be read. From the sd file the 13C and a 1H spectra are read. nmrshiftdb2 offers you the possibility to predict spectra of all type. The quality of the prediction depends on the database content, so for types with only a few spectra it is unlikely you get a good prediction. To do so, you need to enter your molecule (about entering molecules see Submitting Spectra) and to submit it. Your structures are collected in the structures history. You can also choose (via "Use measured and/or calulated spectra") to use only measured or calculated spectra (checking none of both options has the same effect as checking both). There might be errors in the calculated spectra sometimes, so measured only is the default choice. You are then presented with the predicted spectrum in a table and a picture. If you hover over the table, an atom and its shift are marked. For spectrum types other than 1H the predictions are based on HOSE codes. These basically describe the neighbourhood of an atom in concentric rings ("spheres"). If two molecules have a similar neighbourhood, they will have the same HOSE code for a certain number of spheres. The number of spheres used is also given. The more spheres used, the better the prediction. Up to 6 spheres may be used; if less are actually used this means that there are no atoms with a higher number of identical spheres in the database. Since the predictions are won of similar atoms in the molecules in nmrshiftdb2, there can be multiple values for one molecule. What you get in the shift column of the table is actually the average of these. The statistics column gives also the minimum, the maximum, the median and the standard deviation of all values. The checkbox "Use 3D hose codes" is checked by default. If this is used, a stereochemically extended HOSE code is used. This will lead to stereochemistry aware predictions with respect to double bond configurations and chiral centres. Figure 1 and Figure 2 give examples for this. Also certain diastereotopic carbons are respected (see Figure 3). Note that two requirements must be fulfilled for a stereochemistry aware prediction: you must specify the input with wedge bonds (3d coordinates, "perspective drawings" etc. will not be considered) and a matching neighbourhood must exist in the database. If this is not the case, prediction falls back to normal HOSE codes. For proton prediction apart from HOSE code, a prediction based on 3D descriptors and Support Vector Machines ("SVMs") is offered ("1H svm" in the drop down menu). This can give better results than HOSE codes, but should be treated with care. See this paper for details. A prediction based on a deep learning procedure is available for 13C and 1H predictions ("1H NN" and "13C NN"). A publication is in preparation. In order to enable quality control by users, we have a rating system working. Every spectrum has a rating from 1 to 10, default is 10. Every registered user can "vote" on a spectrum. The rating of a spectrum is the average of all ratings, including the default rating. There are two important thresholds: If the rating is less than 5, it is no longer used for predictions, is not included in the spectrum search and does not count for the current usage statistics. If the rating is below 3, it needs an extra click to view it. Spectra never get deleted because of ratings. If the rating of a spectrum falls below the threshold, the administrators are notified and can check the spectrum. nmrshiftdb2 automatically collects the last 20 structures you painted in search by structure, predict or submit. You can load one of these structeres at any time by clicking the "Import from structures history" button next to a sketcher. You get a window where you can choose a structure. Clicking "Import" imports this and closes the import window. The collection of structures is session-based, which means that structures are lost if you log in, log out or close the browser window. This is not true for registered and logged in users - they will find their old structures history if they log in again. nmrshiftdb2 can work as a user and order administration system for NMR labs. Note that the lab system is an additional feature which must be installed in-house. The standard nmrshiftdb2 servers do not offer it. The concept of the lab system is centered around two roles, operator and user, and the concept of an order. Users can assign themself membership of a labgroup, either as operator or user. They need to be approved by the group leader. Once this is done, a user can submit orders and a operator can work on them. A user, when logged in, has an additional tab "NMR lab administration". Here, he has a form which he needs to fill out to submit an order. There are three possible ways to handle an order: an operator: This means a lab operator will handle the order. After the submission, it will show up in the list of open orders. If a operator has dealt with this, it goes to the fullfilled orders list. The user can click on a fullfilled order and can download the raw data files then. He can also start an assignment of peaks here, which is done via the normal NRMShiftDB input. By the user himself: Here the user needs to do the measurement. When he clicks on the order on the open orders list, he can browse through the data files and attach them to the order. Once this is done, the procedure is the same as above. By the user via a sample changer: Here the user needs to put his probe into the sample changer and it will be measured during the next night. The data files get attached automatically and the order goes to the fullfilled orders list.
The user can delete (=cancel) orders which have not yet been processed. Once an order is processed (i. e. has files assigned to it) it can not be deleted, because it is needed for statistcs. It stays in the "done orders" till a peak assignment is done. If the user does not want to do a peak assignment, he can view the order by clicking it and choose "I do not want to assign the spectra", having checked "confirm" before, which will take the order from the list as well. If the user wants to see the the orders which either have a peak assignment or where the user decided not to do it, he can choose "Also show orders already assigned" and press "Change dates/show alreayd assigned". With "I want to measure this sample again" the order is marked as not to be assigned and the user gets the submit order form, with the data of the order filled in. There is also a direct link to the Faces Scheduling System on the page, which only works, if user name and password are the same in Faces as in nmrshiftdb2 An operator, when logged in, also has the "NMR lab administration" tab. He sees a list of open orders. When clicking on one, he can assign raw data files to it and declare the order to be finished. The operator can also delete "operator"-orders. It will then disappear from the list. The worker can also view orders, which have been done by users or sample changer for a certain period. These orders can be made an "operator"-order by the operator. This is helpful in case the automatic assignment does not work (e. g. misspelled ID), it can then be processed manually. Furthermore, operators can generate statistics in pdf format. One operator is designated as group leader; he can admit users to the group and administer the spectrometers and experiments. In order to create a new labgroup on a machine (which must be under the administration of the group. which wants to use the feature), an entry into the labgroup table must be done manually. Further lab system topics can be found in the nmrshiftdb2 wiki. There are some miscellaneous services offered by nmrshiftdb2. These are listed here: |
|
| |
|
|
|
|